This is a read-only mirror of pymolwiki.org
Difference between revisions of "BbPlane"
Jump to navigation
Jump to search
(fix H selection) |
m (10 revisions) |
||
| (5 intermediate revisions by 2 users not shown) | |||
| Line 1: | Line 1: | ||
| + | {{Infobox script-repo | ||
| + | |type = module | ||
| + | |filename = bbPlane.py | ||
| + | |author = [[User:Inchoate|Jason Vertrees]], [[User:Bell|Blaine Bell]] and [[User:Speleo3|Thomas Holder]] | ||
| + | |license = MIT | ||
| + | }} | ||
| + | |||
This script will draw a CGO plane between the backbone atoms of two neighboring residues. This is to show the planarity of the atoms. The image style this is meant to represent can be found many places, like "Introduction to Protein Structure" by Branden and Tooze (2nd ed. pp. 8). | This script will draw a CGO plane between the backbone atoms of two neighboring residues. This is to show the planarity of the atoms. The image style this is meant to represent can be found many places, like "Introduction to Protein Structure" by Branden and Tooze (2nd ed. pp. 8). | ||
| Line 14: | Line 21: | ||
# make planes for residues 4-9 | # make planes for residues 4-9 | ||
bbPlane i. 4-10 | bbPlane i. 4-10 | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
</source> | </source> | ||
| Line 121: | Line 26: | ||
[[Category:Math_Scripts]] | [[Category:Math_Scripts]] | ||
[[Category:Structural_Biology_Scripts]] | [[Category:Structural_Biology_Scripts]] | ||
| + | [[Category:Pymol-script-repo]] | ||
Latest revision as of 01:31, 28 March 2014
| Type | Python Module |
|---|---|
| Download | bbPlane.py |
| Author(s) | Jason Vertrees, Blaine Bell and Thomas Holder |
| License | MIT |
| This code has been put under version control in the project Pymol-script-repo | |
This script will draw a CGO plane between the backbone atoms of two neighboring residues. This is to show the planarity of the atoms. The image style this is meant to represent can be found many places, like "Introduction to Protein Structure" by Branden and Tooze (2nd ed. pp. 8).
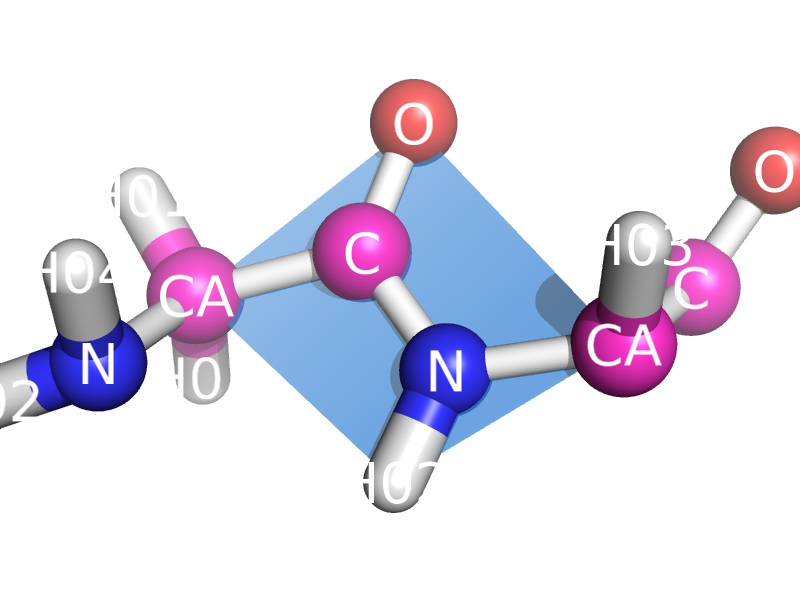
Close up of planar atoms
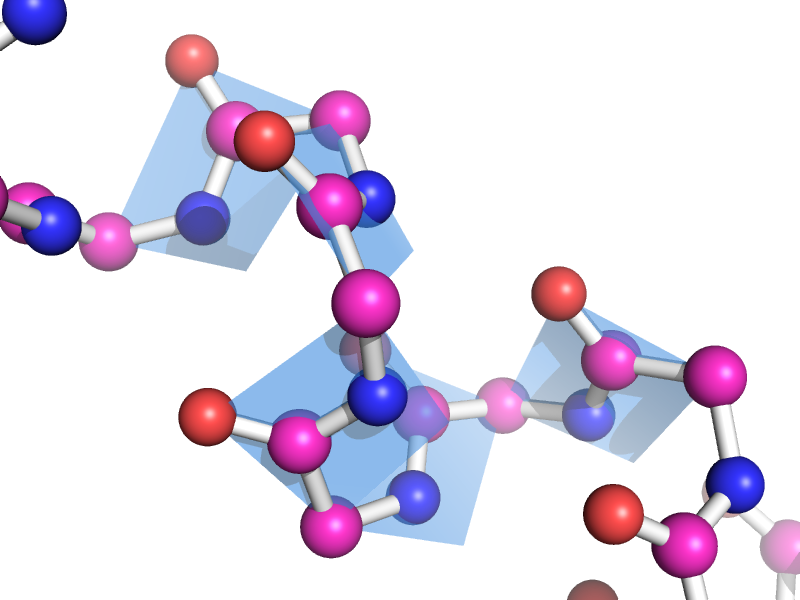
A few more

Global view



Examples
# download the source and save as bbPlane.py
run bbPlane.py
fetch 1cll
# make planes for residues 4-9
bbPlane i. 4-10