This is a read-only mirror of pymolwiki.org
Difference between revisions of "BbPlane"
Jump to navigation
Jump to search
(multistate support, simplification of vertex ordering) |
(No difference)
|
Revision as of 14:53, 26 January 2012
This script will draw a CGO plane between the backbone atoms of two neighboring residues. This is to show the planarity of the atoms. The image style this is meant to represent can be found many places, like "Introduction to Protein Structure" by Branden and Tooze (2nd ed. pp. 8).
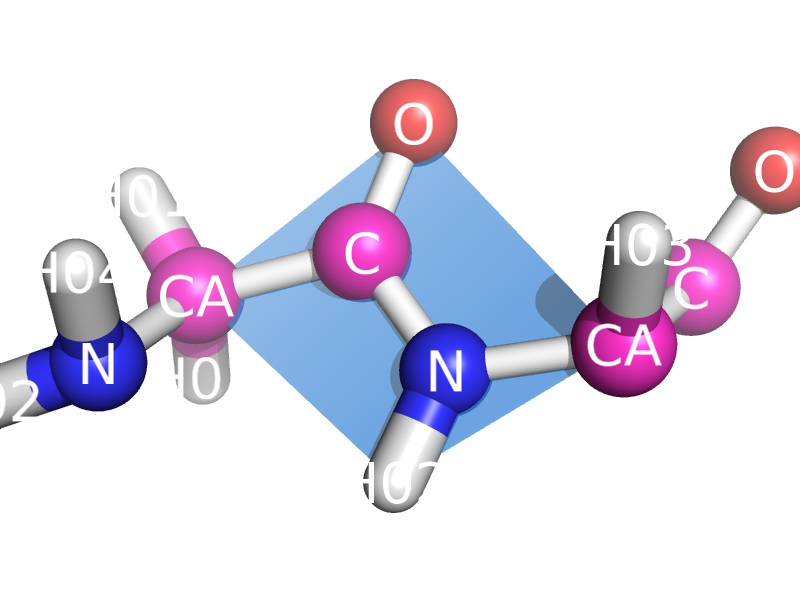
Close up of planar atoms
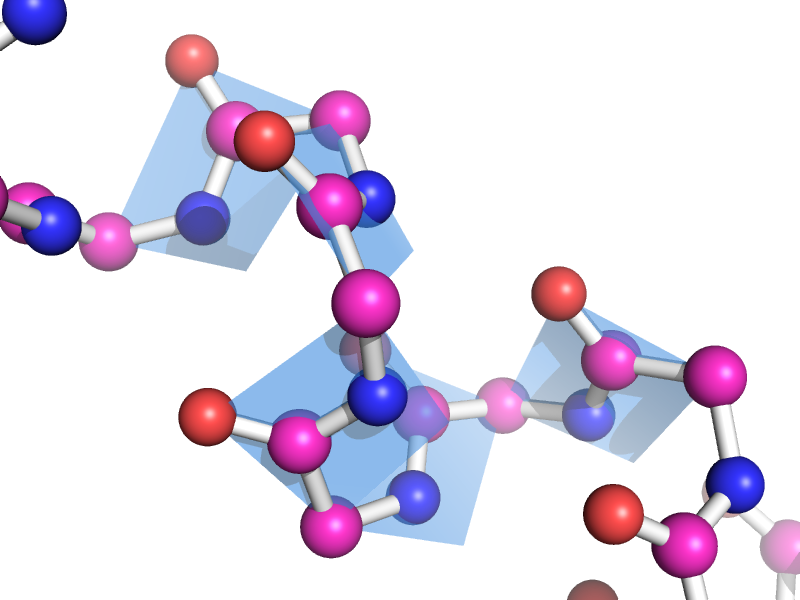
A few more

Global view



Examples
# download the source and save as bbPlane.py
run bbPlane.py
fetch 1cll
# make planes for residues 4-9
bbPlane i. 4-10
The Source
#
# -- bbPlane.py - draws a CGO plane across the backbone atoms of
# neighboring amino acids
#
# Author: Jason Vertrees, 06/2010
# Modified by Thomas Holder, 06/2010
# Modified by Blaine Bell, 08/2011
# Copyright (C) Schrodinger
# Open Source License: MIT
#
from pymol.cgo import * # get constants
from pymol import cmd, stored
from chempy import cpv
def bbPlane(objSel='(all)', color='white', transp=0.0, state=1, name=None, quiet=1):
"""
DESCRIPTION
Draws a plane across the backbone for a selection
ARGUMENTS
objSel = string: protein object or selection {default: (all)}
color = string: color name or number {default: white}
transp = float: transparency component (0.0--1.0) {default: 0.0}
state = integer: object state, 0 for all states {default: 1}
NOTES
You need to pass in an object or selection with at least two
amino acids. The plane spans CA_i, O_i, N-H_(i+1), and CA_(i+1)
"""
# format input
transp = float(transp)
state, quiet = int(state), int(quiet)
if name is None:
name = cmd.get_unused_name("backbonePlane")
if state < 0:
state = cmd.get_state()
elif state == 0:
for state in range(1, cmd.count_states(objSel)+1):
bbPlane(objSel, color, transp, state, name, quiet)
return
stored.AAs = []
coords = dict()
# need hydrogens on peptide nitrogen
cmd.h_add('(%s) and n. N' % objSel)
# get the list of residue ids
for obj in cmd.get_object_list(objSel):
sel = obj + " and (" + objSel + ")"
for a in cmd.get_model(sel + " and n. CA", state).atom:
key = '/%s/%s/%s/%s' % (obj,a.segi,a.chain,a.resi)
stored.AAs.append(key)
coords[key] = [a.coord,None,None]
for a in cmd.get_model(sel + " and n. O", state).atom:
key = '/%s/%s/%s/%s' % (obj,a.segi,a.chain,a.resi)
if key in coords:
coords[key][1] = a.coord
for a in cmd.get_model(sel + " and ((n. N extend 1 and e. H) or (r. PRO and n. CD))", state).atom:
key = '/%s/%s/%s/%s' % (obj,a.segi,a.chain,a.resi)
if key in coords:
coords[key][2] = a.coord
# need at least two amino acids
if len(stored.AAs) <= 1:
print "ERROR: Please provide at least two amino acids, the alpha-carbon on the 2nd is needed."
return
# prepare the cgo
obj = [
BEGIN, TRIANGLES,
COLOR,
]
obj.extend(cmd.get_color_tuple(color))
for res in range(0, len(stored.AAs)-1):
curIdx, nextIdx = str(stored.AAs[res]), str(stored.AAs[res+1])
# populate the position array
pos = [coords[curIdx][0], coords[curIdx][1], coords[nextIdx][2], coords[nextIdx][0]]
# if the data are incomplete for any residues, ignore
if None in pos:
print 'peptide bond %s -> %s incomplete' % (curIdx, nextIdx)
continue
if cpv.distance(pos[0], pos[3]) > 4.0:
print '%s and %s not adjacent' % (curIdx, nextIdx)
continue
# need to order vertices to generate correct triangles for plane
# modified/added by B.Bell 8/18/2011
# modified by Thomas Holder 2012
if cpv.dot_product(cpv.sub(pos[0], pos[1]), cpv.sub(pos[2], pos[3])) < 0:
vorder = [0,1,2,2,3,0]
else:
vorder = [0,1,2,3,2,1]
# fill in the vertex data for the triangles;
for i in vorder:
obj.append(VERTEX)
obj.extend(pos[i])
# finish the CGO
obj.append(END)
# update the UI
cmd.load_cgo(obj, name, state)
cmd.set("cgo_transparency", transp, name)
cmd.extend("bbPlane", bbPlane)