This is a read-only mirror of pymolwiki.org
Align
DESCRIPTION
align performs a sequence alignment followed by a structural alignment, and then carrys out zero or more cycles of refinement in order to reject structural outliers found during the fit. For comparing proteins with lower sequence identity, an alignment program like, Cealign might be a better choice.
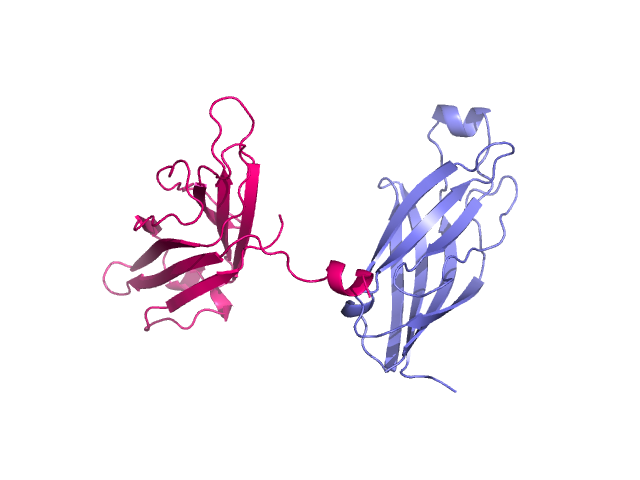
Two unaligned proteins
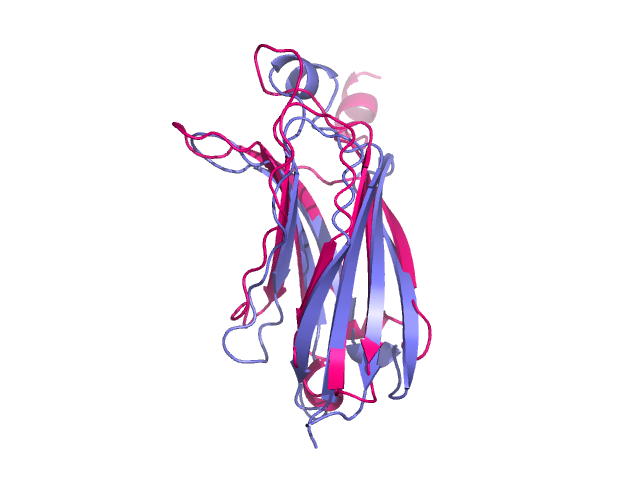
Two proteins after structure alignment


Algorithm Details
align does a BLAST-like BLOSUM62-weighted dynamic programming sequence alignment followed by a series of refinement cycles intended to improve the fit by eliminating pairing with high relative variability (e.g. >2 standard deviations from the cycle's mean deviance).
Your can write the final alignment to a file see save.
Super!
PyMOL now has another command -- super. Super allows for much more robust alignments. It's fast, and under testing, does MUCH better than the original align command.
USAGE
align (source), (target) [,cutoff [,cycles [,gap [,extend \
[,skip [,object [,matrix [, quiet ]]]]]]]]
PYMOL API
cmd.align( string source, string target, float cutoff=2.0,
int cycles=2, float gap=-10.0, float extend=-0.5,
int skip=0, string object=None, string matrix="BLOSUM62",
int quiet=1 )
EXAMPLES
align prot1////CA, prot2, object=alignment
NOTE
- If object is not None, then align will create an object which indicates which atoms were paired between the two structures
- Important note: the molecules you want to align need to be in two different objects. Else, PyMol will answer with a rather cryptic error:
ExecutiveAlign: invalid selections for alignment.
You can skirt this problem by making a temporary object and aligning your original to the copy.
- Sometimes Align may appear to give a mediocre fit. This is not due to any shortcoming of the algorithm or Pymol for that matter. This usually happens if one or more of the objects, that you are trying to align, have multiple states. For instance, certain PDB files may contain multiple structures/ensembles of the same protein. This is especially true for PDB files containing NMR structures. The workaround in such a situation is to use this workflow (provided by Warren - thanks!):
set all_states, on
intra_fit <your_structure_1>
intra_fit <your_structure_2>
align <your_structure_1>////CA, <your_structure_2>////CA
SEE ALSO
fit, rms, rms_cur, intra_rms, intra_rms_cur, pair_fit, intra_fit, Kabsch, Cealign.