This is a read-only mirror of pymolwiki.org
Difference between revisions of "Ccp4 ncont"
Jump to navigation
Jump to search
| Line 1: | Line 1: | ||
| − | |||
| − | |||
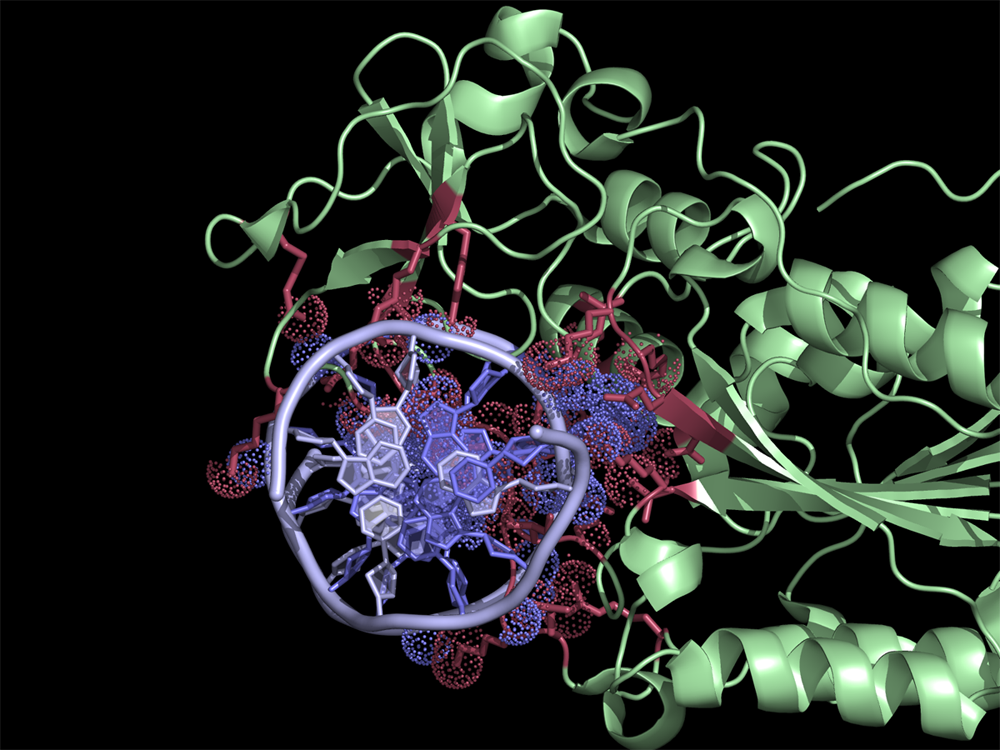
[[File:HhaExample.png|thumb|300px|right|Interface residues (at cutoff <4A) in the 2c7r.pdb were found using NCONT. Usage of ContactsNCONT script in PyMOL allows easy selection of residues and atoms listed in ncont.log file. Interacting protein and DNA residues are colored in red and slate, respectively. Atoms in contact are shown in dots.]] | [[File:HhaExample.png|thumb|300px|right|Interface residues (at cutoff <4A) in the 2c7r.pdb were found using NCONT. Usage of ContactsNCONT script in PyMOL allows easy selection of residues and atoms listed in ncont.log file. Interacting protein and DNA residues are colored in red and slate, respectively. Atoms in contact are shown in dots.]] | ||
Revision as of 14:47, 20 November 2009

Interface residues (at cutoff <4A) in the 2c7r.pdb were found using NCONT. Usage of ContactsNCONT script in PyMOL allows easy selection of residues and atoms listed in ncont.log file. Interacting protein and DNA residues are colored in red and slate, respectively. Atoms in contact are shown in dots.
Overview
The script selects residues and atoms from the list of the contacts found by NCONT from CCP4 Program Suite (NCONT analyses contacts between subsets of atoms in a PDB file). First, we run NCONT on our pdb file to find interface residues. Then by using the ContactsNCONT script in PyMOL we select listed residues and atoms separately. This generates two selections (atoms and residues) for each interacting chain, allowing quick manipulation of (sometimes) extensive lists in NCONT log file.
Usage
selectContacts( contactsfile, selName1 = "source", selName2 = "target" )
Examples
First use NCONT to find interface residues/atoms in the same pdb file. Once you have ncont.log file proceed to PyMOL. Make sure you've run the ContactsNCONT script first. fetch 2c7r selectContacts ncont.log, selName1=prot, selName2=dna
The Code
import re
def parseContacts( f ):
# /1/B/ 282(PHE). / CE1[ C]: /1/E/ 706(GLN). / O [ O]: 3.32
conParser = re.compile("\s*/(\d+)/([A-Z])/\s*(\d+).*?/\s*([A-Z0-9]*).*?:")
mode = 0
s1 = []
s2 = []
pairs = []
for line in f:
if mode == 0:
if line.strip().startswith("SOURCE ATOMS"):
mode = 1
elif mode == 1:
mode = 2
elif mode == 2:
matches = conParser.findall(line)
if len(matches) == 0:
return (s1, s2, pairs)
if len(matches) == 2:
s1.append(matches[0])
s2.append(matches[1])
elif len(matches) == 1:
s2.append(matches[0])
pairs.append((len(s1)-1, len(s2)-1))
else:
print "Unknown mode", mode
def selectContacts( contactsfile, selName1 = "source", selName2 = "target" ):
"""
selectContacts -- parses CCP4 NCONT log file and selects residues and atoms from the list of the contacts found.
PARAMS
contactsfile
filename of the CCP4 NCONT contacts log file
selName1
the name prefix for the _res and _atom selections returned for the
source set of chain
selName2
the name prefix for the _res and _atom selections returned for the
target set of chain
RETURNS
* 2 selections of interface residues and atoms for each chain are created and named
depending on what you passed into selName1 and selName2
AUTHOR:
Gerhard Reitmayr and Dalia Daujotyte, 2009.
"""
# read and parse contacts file into two lists of contact atoms and contact pair list
s1, s2, pairs = parseContacts(open(contactsfile))
# create a selection for the first contact list
resName = selName1 + "_res"
atomName = selName1 + "_atom"
cmd.select(resName, None)
cmd.select(atomName, None)
for (thing, chain, residue, atom) in s1:
cmd.select( resName, resName + " or " + chain+"/"+residue+"/")
cmd.select( atomName, atomName + " or " + chain+"/"+residue+"/"+atom)
# create a selection for the second contact list
resName = selName2 + "_res"
atomName = selName2 + "_atom"
cmd.select(resName, None)
cmd.select(atomName, None)
for (thing, chain, residue, atom) in s2:
cmd.select( resName, resName + " or " + chain+"/"+residue+"/")
cmd.select( atomName, atomName + " or " + chain+"/"+residue+"/"+atom)
cmd.extend("selectContacts", selectContacts)