This is a read-only mirror of pymolwiki.org
Difference between revisions of "Rotamer Toggle"
m (47 revisions) |
|||
| (44 intermediate revisions by 3 users not shown) | |||
| Line 1: | Line 1: | ||
===DESCRIPTION=== | ===DESCRIPTION=== | ||
| − | Backbone-Dependent Rotamer library (Dunbrack, Cohen see ref) is imported into pymol giving access to this information. There are a number of different ways to use the data, I've only implemented a few as well as added extra functions that seemed useful. | + | Backbone-Dependent Rotamer library (Dunbrack, Cohen ; see ref) is imported into pymol giving access to this information. There are a number of different ways to use the data, I've only implemented a few as well as added extra functions that seemed useful. |
| + | *Rotamer Menu - an added menu into menu.py, which displays the most common rotamers for the given(clicked) residue; you can also set the residue any of the common rotamers as well | ||
*colorRotamers - color rotamers by closest matching rotamer angles from database; i.e. color by how common each rotamer of selection, blue - red (least to most common). | *colorRotamers - color rotamers by closest matching rotamer angles from database; i.e. color by how common each rotamer of selection, blue - red (least to most common). | ||
| − | |||
*set_rotamer - routine called by above menu, but can be called manually to set a specific residues side-chain angles | *set_rotamer - routine called by above menu, but can be called manually to set a specific residues side-chain angles | ||
| − | *set_phipsi - set all phi,psi angles of given selection to given angles (useful for creating secondary structures) | + | *set_phipsi - set all phi,psi angles of given selection to given angles (useful for creating secondary structures) |
| + | *createRotamerPDBs - create pdb for each rotamer of given selection ; filter by rotamer-probability | ||
| + | |||
| + | ===IMAGES=== | ||
| + | <gallery> | ||
| + | Image:RotamerMenu.png|Rotamer Menu for a GLN residue | ||
| + | Image:GLURotamerComparison5.png|Rotamer Comparison of crystal structure and most common for GLU; just as an example | ||
| + | </gallery> | ||
| + | |||
| + | Print out while selecting most common rotamer from above-left image (GLN residue): | ||
| + | Given GLN:40 PHI,PSI (-171.626373291,-96.0500335693) : bin (-170,-100) | ||
| + | CHIs: [179.18069458007812, 72.539344787597656, -47.217315673828125] | ||
| + | Setting Chi1 to -176.9 | ||
| + | Setting Chi2 to 177.4 | ||
| + | Setting Chi3 to 0.7 | ||
| + | |||
| + | ===SETUP=== | ||
| + | run "rotamers.py" and use functions from commandline. | ||
| + | |||
| + | or | ||
| − | To setup a rotamer menu inside the residue menu: | + | To setup a rotamer menu inside the residue menu (default windows pymol installation): |
*copy rotamers.py to C:/Program Files/DeLano Scientific/PyMol/modules/pymol/rotamers.py | *copy rotamers.py to C:/Program Files/DeLano Scientific/PyMol/modules/pymol/rotamers.py | ||
*copy mymenu.py to C:/Program Files/DeLano Scientific/PyMol/modules/pymol/menu.py (WARNING : overwrites default menu.py - use at your own risk) | *copy mymenu.py to C:/Program Files/DeLano Scientific/PyMol/modules/pymol/menu.py (WARNING : overwrites default menu.py - use at your own risk) | ||
*copy bbdep02.May.sortlib to C:/Program Files/DeLano Scientific/PyMol/modules/pymol/bbdep02.May.sortlib (or newer version of sorted bbdep) | *copy bbdep02.May.sortlib to C:/Program Files/DeLano Scientific/PyMol/modules/pymol/bbdep02.May.sortlib (or newer version of sorted bbdep) | ||
| − | This is only one possible way to do this, I am sure there are many others. | + | This is only one possible way to do this, I am sure there are many others. I'm not going to post the bbdep, but there is a link in the References section to Dunbrack's download page (get the "sorted" lib) |
| + | |||
| + | ===NOTES / STATUS=== | ||
| + | *Tested on Pymolv0.97, Windows platform, Red Hat Linux 9.0 and Fedora Core 4. Will test v0.98 and MacOSX later on. | ||
| + | *The way it's setup now, when you import rotamers , it will automatically read-in the rotamer database; this may not be what you want. | ||
| + | *Post problems in the discussion page, on 'my talk' page or just email me : dwkulp@mail.med.upenn.edu | ||
| + | |||
| + | TASKS TODO: | ||
| + | *Rotamer Movie, using mset, etc create movie to watch cycle through rotamers | ||
| + | *Code could be organized a bit better; due to time constraints this is good for now.. | ||
| + | |||
| + | TASKS DONE: | ||
| + | *Store crystal structure in rotamer menu, so you can go back to original orientation | ||
===USAGE=== | ===USAGE=== | ||
| Line 16: | Line 47: | ||
set_rotamer selection, chi1_angle [,chi2_angle] [,chi3_angle] [,chi4_angle] | set_rotamer selection, chi1_angle [,chi2_angle] [,chi3_angle] [,chi4_angle] | ||
set_phipsi selection phi_angle, psi_angle | set_phipsi selection phi_angle, psi_angle | ||
| − | + | createRotamerPBDs selection [,ncutoff] [,pcutoff] [,prefix] | |
| − | |||
| − | |||
| − | |||
| − | |||
===EXAMPLES=== | ===EXAMPLES=== | ||
| − | + | colorRotamers chain A | |
| − | + | set_rotamer resi 40, -60,-40 (only set chi1,chi2 angles) | |
| + | set_phipsi resi 10-40, -60,-60 (create an alpha-helical-like section) | ||
| + | createRotamerPDBs resi 10-12, ncutoff=3 (create 9 PDBs; each with one of the 3 most probable rotamers for resi 10,11,12) | ||
| + | createRotamerPDBs resi 14, pcutoff=0.4 (create a pdb file for each rotamer of residue 14 with probablity > 0.4) | ||
| − | |||
| − | |||
===REFERENCES=== | ===REFERENCES=== | ||
Dunbrack and Cohen. Protein Science 1997 | Dunbrack and Cohen. Protein Science 1997 | ||
[http://dunbrack.fccc.edu/bbdep/index.php Dunbrack Lab Page (Contains backbone-dependent library)] | [http://dunbrack.fccc.edu/bbdep/index.php Dunbrack Lab Page (Contains backbone-dependent library)] | ||
| + | |||
| + | ===SCRIPTS (Rotamers.py ; MyMenu.py)=== | ||
| + | Rotamers.py | ||
| + | <source lang="python"> | ||
| + | ################################################################## | ||
| + | # File: Rotamers.py | ||
| + | # Author: Dan Kulp | ||
| + | # Creation Date: 6/8/05 | ||
| + | # Contact: dwkulp@mail.med.upenn.edu | ||
| + | # | ||
| + | # Notes: | ||
| + | # Incorporation of Rotamer library | ||
| + | # readRotLib() - fills rotdat; | ||
| + | # indexed by "RES:PHI_BIN:PSI_BIN". | ||
| + | # | ||
| + | # Three main functions: | ||
| + | # 1. colorRotamers - colors according | ||
| + | # to rotamer probablitity | ||
| + | # 2. getBins(sel) | ||
| + | # phi,psi bin for rotamer | ||
| + | # 3. set_rotamer - set a side-chain | ||
| + | # to a specific rotamer | ||
| + | # | ||
| + | # To setup a rotamer menu in the | ||
| + | # right click, under "Residue" | ||
| + | # 1. cp mymenu.py modules/pymol/menu.py | ||
| + | # 2. cp rotamers.py modules/pymol/rotamers.py (update ROTLIB) | ||
| + | # | ||
| + | # Requirements: | ||
| + | # set ROTLIB to path for rotamer library | ||
| + | # Reference: | ||
| + | # Dunbrack and Cohen. Protein Science 1997 | ||
| + | #################################################################### | ||
| + | |||
| + | import colorsys,sys | ||
| + | import editing | ||
| + | import os | ||
| + | import cmd | ||
| + | import math | ||
| + | |||
| + | # Path for library | ||
| + | ROTLIB=os.environ['PYMOL_PATH']+"/modules/pymol/bbdep02.May.sortlib" | ||
| + | |||
| + | # Place for library in memory.. | ||
| + | rotdat = {} | ||
| + | |||
| + | def readRotLib(): | ||
| + | # Column indexes in rotamer library.. | ||
| + | RES = 0 | ||
| + | PHI = 1 | ||
| + | PSI = 2 | ||
| + | PROB = 8 | ||
| + | CHI1 = 9 | ||
| + | CHI2 = 10 | ||
| + | CHI3 = 11 | ||
| + | CHI4 = 12 | ||
| + | |||
| + | if os.path.exists(ROTLIB): | ||
| + | print "File exists: "+ROTLIB | ||
| + | input = open(ROTLIB, 'r') | ||
| + | for line in input: | ||
| + | |||
| + | # Parse by whitespace (I believe format is white space and not fixed-width columns) | ||
| + | dat = line.split() | ||
| + | |||
| + | # Add to rotamer library in memory : | ||
| + | # key format RES:PHI_BIN:PSI_BIN | ||
| + | # value format PROB, CHI1, CHI2, CHI3, CHI4 | ||
| + | key=dat[RES]+":"+dat[PHI]+":"+dat[PSI] | ||
| + | if key in rotdat: | ||
| + | rotdat[key].append([ dat[PROB], dat[CHI1], dat[CHI2], dat[CHI3], dat[CHI4] ]) | ||
| + | else: | ||
| + | rotdat[key] = [ [ dat[PROB], dat[CHI1], dat[CHI2], dat[CHI3], dat[CHI4] ] ] | ||
| + | |||
| + | |||
| + | else: | ||
| + | print "Couldn't find Rotamer library" | ||
| + | |||
| + | |||
| + | # Atoms for each side-chain angle for each residue | ||
| + | CHIS = {} | ||
| + | CHIS["ARG"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","CD" ], | ||
| + | ["CB","CG","CD","NE" ], | ||
| + | ["CG","CD","NE","CZ" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["ASN"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","OD2" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["ASP"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","OD1" ] | ||
| + | ] | ||
| + | CHIS["CYS"] = [ ["N","CA","CB","SG" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["GLN"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","CD" ], | ||
| + | ["CB","CG","CD","OE1"] | ||
| + | ] | ||
| + | |||
| + | CHIS["GLU"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","CD" ], | ||
| + | ["CB","CG","CD","OE1"] | ||
| + | ] | ||
| + | |||
| + | CHIS["HIS"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","ND1"] | ||
| + | ] | ||
| + | |||
| + | CHIS["ILE"] = [ ["N","CA","CB","CG1" ], | ||
| + | ["CA","CB","CG1","CD1" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["LEU"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","CD1" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["LYS"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","CD" ], | ||
| + | ["CB","CG","CD","CE"], | ||
| + | ["CG","CD","CE","NZ"] | ||
| + | ] | ||
| + | |||
| + | CHIS["MET"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","SD" ], | ||
| + | ["CB","CG","SD","CE"] | ||
| + | ] | ||
| + | |||
| + | CHIS["PHE"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","CD1" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["PRO"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","CD" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["SER"] = [ ["N","CA","CB","OG" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["THR"] = [ ["N","CA","CB","OG1" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["TRP"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","CD1"] | ||
| + | ] | ||
| + | |||
| + | CHIS["TYR"] = [ ["N","CA","CB","CG" ], | ||
| + | ["CA","CB","CG","CD1" ] | ||
| + | ] | ||
| + | |||
| + | CHIS["VAL"] = [ ["N","CA","CB","CG1" ] | ||
| + | ] | ||
| + | |||
| + | # Color Rotamer by side-chain angle position | ||
| + | # 'bin' side-chain angles into closest | ||
| + | def colorRotamers(sel): | ||
| + | doRotamers(sel) | ||
| + | |||
| + | # Utility function, to set phi,psi angles for a given selection | ||
| + | # Note: Cartoon, Ribbon functionality will not display correctly after this | ||
| + | def set_phipsi(sel, phi,psi): | ||
| + | doRotamers(sel,angles=[phi,psi],type="set") | ||
| + | |||
| + | # Set a rotamer, based on a selection, a restype and chi angles | ||
| + | def set_rotamer(sel, chi1, chi2=0,chi3=0,chi4=0): | ||
| + | at = cmd.get_model("byres ("+sel+")").atom[0] | ||
| + | |||
| + | list = [chi1,chi2,chi3,chi4] | ||
| + | for i in range(len(CHIS[at.resn])): | ||
| + | print "Setting Chi"+str(i+1)+" to "+str(list[i]) | ||
| + | editing.set_dihedral(sel + ' and name '+CHIS[at.resn][i][0], | ||
| + | sel + ' and name '+CHIS[at.resn][i][1], | ||
| + | sel + ' and name '+CHIS[at.resn][i][2], | ||
| + | sel + ' and name '+CHIS[at.resn][i][3], str(list[i])) | ||
| + | |||
| + | # Remove some objects that got created | ||
| + | cmd.delete("pk1") | ||
| + | cmd.delete("pk2") | ||
| + | cmd.delete("pkmol") | ||
| + | |||
| + | # Get Phi,Psi bins for given selection | ||
| + | # WARNING: assume selection is single residue (will only return first residue bins) | ||
| + | def getBins(sel): | ||
| + | return doRotamers(sel, type="bins") | ||
| + | |||
| + | # Color Ramp... | ||
| + | def rot_color(vals): | ||
| + | nbins = 10 | ||
| + | vals.sort(key=lambda x:x[1]) | ||
| + | # print "End sort: "+str(len(vals))+" : "+str(nbins) | ||
| + | |||
| + | |||
| + | # Coloring scheme... | ||
| + | j = 0 | ||
| + | rgb = [0.0,0.0,0.0] | ||
| + | sel_str = "" | ||
| + | for i in range(len(vals)): | ||
| + | if int(len(vals)/nbins) == 0 or i % int(len(vals)/nbins) == 0: | ||
| + | hsv = (colorsys.TWO_THIRD - colorsys.TWO_THIRD * float(j) / (nbins-1), 1.0, 1.0) | ||
| + | |||
| + | #convert to rgb and append to color list | ||
| + | rgb = colorsys.hsv_to_rgb(hsv[0],hsv[1],hsv[2]) | ||
| + | if j < nbins-1: | ||
| + | j += 1 | ||
| + | |||
| + | cmd.set_color("RotProbColor"+str(i), rgb) | ||
| + | cmd.color("RotProbColor"+str(i), str(vals[i][0])) | ||
| + | |||
| + | |||
| + | # Main function | ||
| + | def doRotamers(sel,angles=[], type="color"): | ||
| + | |||
| + | # Read in Rotamer library if not already done | ||
| + | if len(rotdat) == 0: | ||
| + | readRotLib() | ||
| + | |||
| + | # Set up some variables.. | ||
| + | residues = ['dummy'] # Keep track of residues already done | ||
| + | probs = [] # probability of each residue conformation | ||
| + | phi = 0 # phi,psi angles of current residue | ||
| + | psi = 0 | ||
| + | |||
| + | # Get atoms from selection | ||
| + | atoms = cmd.get_model("byres ("+sel+")") | ||
| + | |||
| + | # Loop through atoms in selection | ||
| + | for at in atoms.atom: | ||
| + | try: | ||
| + | # Don't process Glycines or Alanines | ||
| + | if not (at.resn == 'GLY' or at.resn == 'ALA'): | ||
| + | if at.chain+":"+at.resn+":"+at.resi not in residues: | ||
| + | residues.append(at.chain+":"+at.resn+":"+at.resi) | ||
| + | |||
| + | # Check for a null chain id (some PDBs contain this) | ||
| + | unit_select = "" | ||
| + | if at.chain != "": | ||
| + | unit_select = "chain "+str(at.chain)+" and " | ||
| + | |||
| + | # Define selections for residue i-1, i and i+1 | ||
| + | residue_def = unit_select+'resi '+str(at.resi) | ||
| + | residue_def_prev = unit_select+'resi '+str(int(at.resi)-1) | ||
| + | residue_def_next = unit_select+'resi '+str(int(at.resi)+1) | ||
| + | |||
| + | # Compute phi/psi angle | ||
| + | |||
| + | phi = cmd.get_dihedral(residue_def_prev+' and name C',residue_def+' and name N',residue_def+' and name CA',residue_def+' and name C') | ||
| + | psi = cmd.get_dihedral(residue_def+' and name N',residue_def+' and name CA',residue_def+' and name C',residue_def_next+' and name N') | ||
| + | if type == "set": | ||
| + | print "Changing "+at.resn+str(at.resi)+" from "+str(phi)+","+str(psi)+" to "+str(angles[0])+","+str(angles[1]) | ||
| + | cmd.set_dihedral(residue_def_prev+' and name C',residue_def+' and name N',residue_def+' and name CA',residue_def+' and name C',angles[0]) | ||
| + | cmd.set_dihedral(residue_def+' and name N',residue_def+' and name CA',residue_def+' and name C',residue_def_next+' and name N', angles[1]) | ||
| + | continue | ||
| + | |||
| + | # Find correct 10x10 degree bin | ||
| + | phi_digit = abs(int(phi)) - abs(int(phi/10)*10) | ||
| + | psi_digit = abs(int(psi)) - abs(int(psi/10)*10) | ||
| + | |||
| + | # Remember sign of phi,psi angles | ||
| + | phi_sign = 1 | ||
| + | if phi < 0: phi_sign = -1 | ||
| + | |||
| + | psi_sign = 1 | ||
| + | if psi < 0: psi_sign = -1 | ||
| + | |||
| + | # Compute phi,psi bins | ||
| + | phi_bin = int(math.floor(abs(phi/10))*10*phi_sign) | ||
| + | if phi_digit >= 5: phi_bin = int(math.ceil(abs(phi/10))*10*phi_sign) | ||
| + | |||
| + | psi_bin = int(math.floor(abs(psi/10))*10*psi_sign) | ||
| + | if psi_digit >= 5: psi_bin = int(math.ceil(abs(psi/10))*10*psi_sign) | ||
| + | |||
| + | print "Given "+at.resn+":"+at.resi+" PHI,PSI ("+str(phi)+","+str(psi)+") : bin ("+str(phi_bin)+","+str(psi_bin)+")" | ||
| + | |||
| + | |||
| + | # Get current chi angle measurements | ||
| + | chi = [] | ||
| + | for i in range(len(CHIS[at.resn])): | ||
| + | chi.append(cmd.get_dihedral(residue_def + ' and name '+CHIS[at.resn][i][0], | ||
| + | residue_def + ' and name '+CHIS[at.resn][i][1], | ||
| + | residue_def + ' and name '+CHIS[at.resn][i][2], | ||
| + | residue_def + ' and name '+CHIS[at.resn][i][3])) | ||
| + | print "CHIs: "+str(chi) | ||
| + | if type == 'bins': | ||
| + | return [at.resn, phi_bin,psi_bin] | ||
| + | |||
| + | # Compute probabilities for given chi angles | ||
| + | prob = 0 | ||
| + | prob_box = 22 | ||
| + | for item in range(len(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)])): | ||
| + | print "Rotamer from db: "+str(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item]) | ||
| + | if chi[0]: | ||
| + | if chi[0] >= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][1]) - (prob_box/2) and \ | ||
| + | chi[0] <= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][1]) + (prob_box/2): | ||
| + | if len(chi) == 1: | ||
| + | prob = rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][0] | ||
| + | break | ||
| + | if chi[1] >= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][2]) - (prob_box/2) and \ | ||
| + | float(chi[1] <= rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][2]) + (prob_box/2): | ||
| + | if len(chi) == 2: | ||
| + | prob = rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][0] | ||
| + | break | ||
| + | if chi[2] >= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][3]) - (prob_box/2) and \ | ||
| + | float(chi[2] <= rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][3]) + (prob_box/2): | ||
| + | if len(chi) == 3: | ||
| + | prob = rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][0] | ||
| + | break | ||
| + | if chi[3] >= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][4]) - (prob_box/2) and \ | ||
| + | float(chi[3] <= rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][4]) + (prob_box/2): | ||
| + | prob = rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][0] | ||
| + | break | ||
| + | |||
| + | |||
| + | print "PROB OF ROTAMER: "+str(prob) | ||
| + | print "---------------------------" | ||
| + | probs.append([residue_def, prob]) | ||
| + | |||
| + | except: | ||
| + | # probs.append([residue_def, -1]) | ||
| + | print "Exception found" | ||
| + | continue | ||
| + | |||
| + | # Color according to rotamer probability | ||
| + | rot_color(probs) | ||
| + | |||
| + | |||
| + | |||
| + | |||
| + | # Create PDB files containing most probable rotamers | ||
| + | def createRotamerPDBs(sel,ncutoff=10,pcutoff=0,prefix="ROTAMER"): | ||
| + | |||
| + | # Get atoms from selection | ||
| + | atoms = cmd.get_model("byres ("+sel+")") | ||
| + | |||
| + | # Set up some variables.. | ||
| + | residues = ['dummy'] # Keep track of residues already done | ||
| + | |||
| + | # Loop through atoms in selection | ||
| + | for at in atoms.atom: | ||
| + | if at.resn in ('GLY','ALA') or "%s:%s:%s" % (at.chain,at.resn,at.resi) in residues: | ||
| + | continue | ||
| + | |||
| + | # Add to residue list (keep track of which ones we've done) | ||
| + | residues.append("%s:%s:%s" % (at.chain,at.resn,at.resi)) | ||
| + | |||
| + | # Check for a null chain id (some PDBs contain this) | ||
| + | unit_select = "" | ||
| + | if not at.chain == "": | ||
| + | unit_select = "chain "+str(at.chain)+" and " | ||
| + | |||
| + | # Define selections for residue | ||
| + | residue_def = unit_select+'resi '+str(at.resi) | ||
| + | |||
| + | # Get bin (phi,psi) definitions for this residue | ||
| + | bin = doRotamers(residue_def, type='bins') | ||
| + | |||
| + | # Store crystal angle | ||
| + | crystal_angles = [0.0,0.0,0.0,0.0] | ||
| + | for angle in range(3): | ||
| + | try: | ||
| + | crystal_angles[angle] = bin[3][angle] | ||
| + | except IndexError: | ||
| + | break | ||
| + | |||
| + | # Retreive list of rotamers for this phi,psi bin + residue type | ||
| + | match_rotamers = rotdat["%s:%s:%s" % (bin[0],str(bin[1]),str(bin[2]))] | ||
| + | |||
| + | count = 0 | ||
| + | for item in range(len(match_rotamers)): | ||
| + | |||
| + | # Store probablity | ||
| + | prob = match_rotamers[item][0] | ||
| + | |||
| + | # Check cutoffs | ||
| + | if float(prob) <= float(pcutoff): | ||
| + | continue | ||
| + | |||
| + | if float(count) >= float(ncutoff): | ||
| + | break | ||
| + | |||
| + | # Increment count | ||
| + | count += 1 | ||
| + | |||
| + | # Output to screen ... | ||
| + | print "Residue %s%s, rotamer %i, prob %s" % (str(at.resn),str(at.resi),int(item),str(prob)) | ||
| + | |||
| + | # Set to new rotamer | ||
| + | set_rotamer(residue_def,match_rotamers[item][1],match_rotamers[item][2],match_rotamers[item][3],match_rotamers[item][4]) | ||
| + | |||
| + | # Store in PDB file | ||
| + | cmd.save("%s_%s%s_%i_%s.pdb" % (prefix,str(at.resn),str(at.resi),int(item),str(prob))) | ||
| + | |||
| + | # Reset crystal angle | ||
| + | set_rotamer(residue_def,crystal_angles[0],crystal_angles[1],crystal_angles[2],crystal_angles[3]) | ||
| + | |||
| + | # Uncommenting this is nice because it loads rotamer library upon startup | ||
| + | # however, it slows the PyMOL loading process a lot | ||
| + | # instead I've put this call into the menuing code.. | ||
| + | # readRotLib() | ||
| + | |||
| + | cmd.extend('set_phipsi',set_phipsi) | ||
| + | cmd.extend('set_rotamer',set_rotamer) | ||
| + | cmd.extend('colorRotamers',colorRotamers) | ||
| + | cmd.extend('createRotamerPDBs',createRotamerPDBs) | ||
| + | </source> | ||
| + | |||
| + | MyMenu.py | ||
| + | Since menu.py is copyrighted I can't post my edited version, but you can create it very simply by adding these two peices of code | ||
| + | |||
| + | 1. In the "pick_option(title,s,object=0)" function of menu.py add the following code after the first "result =" statement | ||
| + | <source lang="python"> | ||
| + | # Edit dwkulp 6/11/05 , add a rotamer menu to residue menu | ||
| + | if title == 'Residue': | ||
| + | result.extend([[ 1, 'rotamers' , rotamer_menu(s)]]) | ||
| + | </source> | ||
| + | |||
| + | 2. At the end of the file add this: | ||
| + | <source lang="python"> | ||
| + | ############################################### | ||
| + | # Dan Kulp | ||
| + | # Added Rotamer list to residue menu.. | ||
| + | # rotamer.py must be importable (I placed it in | ||
| + | # the same directory as menu.py) | ||
| + | ############################################### | ||
| + | |||
| + | import rotamers | ||
| + | |||
| + | |||
| + | def rotamer_menu(s): | ||
| + | # Check for rotamer library being loaded | ||
| + | if not rotamers.rotdat: | ||
| + | rotamers.readRotLib() | ||
| + | # return [ [2, "Must run rotamers.py first",'']] | ||
| + | |||
| + | # Check for valid rotamer residue.. | ||
| + | res = cmd.get_model("byres ("+s+")").atom[0].resn | ||
| + | if not res in rotamers.CHIS.keys(): | ||
| + | return [ [2, "Residue: "+res+" not known sidechain or does not have rotamers", '']] | ||
| + | |||
| + | # Get PHI,PSI bins for rotamer (also prints out current phi,psi, chi1,chi2,chi3,chi4) | ||
| + | bins = rotamers.doRotamers(s,type='bins') | ||
| + | |||
| + | # Add a title to the menu | ||
| + | result = [ [2, bins[0]+' Rotamers in bin ('+str(bins[1])+','+str(bins[2])+')','' ], [1, ':::PROB,CHI1,CHI2,CHI3,CHI4:::','']] | ||
| + | |||
| + | # Grab the entries for this residue and phi,psi bins | ||
| + | match_rotamers = rotamers.rotdat[bins[0]+":"+str(bins[1])+":"+str(bins[2])] | ||
| + | |||
| + | # Set max number of rotamers to display (probably should be somewhere 'higher up' in the code) | ||
| + | max_rotamers = min(10, len(match_rotamers)) | ||
| + | |||
| + | # Create menu entry for each possible rotamer | ||
| + | for item in range(max_rotamers): | ||
| + | result.append( [ 1, str(match_rotamers[item]), 'rotamers.set_rotamer("'+s+'","'\ | ||
| + | +str(match_rotamers[item][1])+'","'\ | ||
| + | +str(match_rotamers[item][2])+'","'\ | ||
| + | +str(match_rotamers[item][3])+'","'\ | ||
| + | +str(match_rotamers[item][4])+'")']) | ||
| + | return result | ||
| + | |||
| + | |||
| + | </source> | ||
| + | |||
[[Category:Script_Library|Rotamer Toggle]] | [[Category:Script_Library|Rotamer Toggle]] | ||
| + | |||
| + | [[Category:Structural_Biology_Scripts|Rotamer Toggle]] | ||
Latest revision as of 03:49, 28 March 2014
DESCRIPTION
Backbone-Dependent Rotamer library (Dunbrack, Cohen ; see ref) is imported into pymol giving access to this information. There are a number of different ways to use the data, I've only implemented a few as well as added extra functions that seemed useful.
- Rotamer Menu - an added menu into menu.py, which displays the most common rotamers for the given(clicked) residue; you can also set the residue any of the common rotamers as well
- colorRotamers - color rotamers by closest matching rotamer angles from database; i.e. color by how common each rotamer of selection, blue - red (least to most common).
- set_rotamer - routine called by above menu, but can be called manually to set a specific residues side-chain angles
- set_phipsi - set all phi,psi angles of given selection to given angles (useful for creating secondary structures)
- createRotamerPDBs - create pdb for each rotamer of given selection ; filter by rotamer-probability
IMAGES
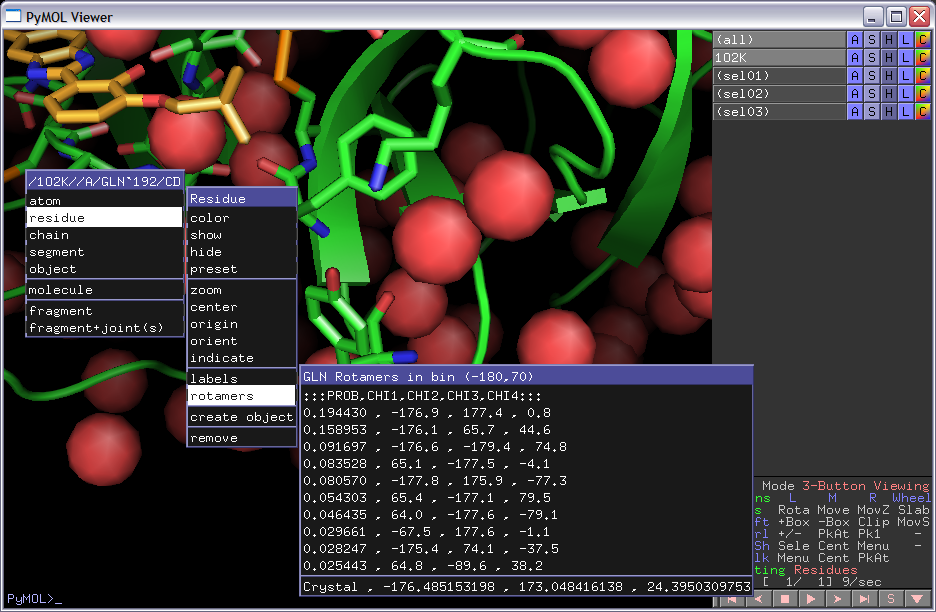
Rotamer Menu for a GLN residue
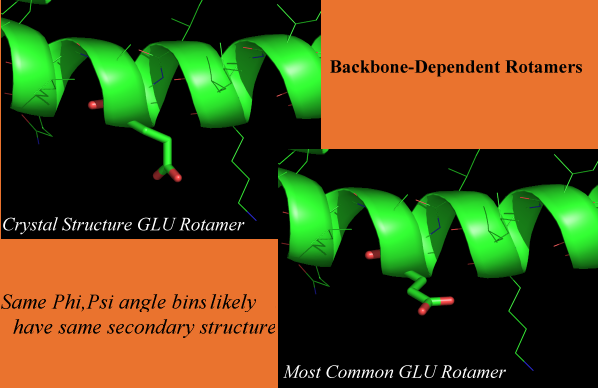
Rotamer Comparison of crystal structure and most common for GLU; just as an example


Print out while selecting most common rotamer from above-left image (GLN residue):
Given GLN:40 PHI,PSI (-171.626373291,-96.0500335693) : bin (-170,-100) CHIs: [179.18069458007812, 72.539344787597656, -47.217315673828125] Setting Chi1 to -176.9 Setting Chi2 to 177.4 Setting Chi3 to 0.7
SETUP
run "rotamers.py" and use functions from commandline.
or
To setup a rotamer menu inside the residue menu (default windows pymol installation):
- copy rotamers.py to C:/Program Files/DeLano Scientific/PyMol/modules/pymol/rotamers.py
- copy mymenu.py to C:/Program Files/DeLano Scientific/PyMol/modules/pymol/menu.py (WARNING : overwrites default menu.py - use at your own risk)
- copy bbdep02.May.sortlib to C:/Program Files/DeLano Scientific/PyMol/modules/pymol/bbdep02.May.sortlib (or newer version of sorted bbdep)
This is only one possible way to do this, I am sure there are many others. I'm not going to post the bbdep, but there is a link in the References section to Dunbrack's download page (get the "sorted" lib)
NOTES / STATUS
- Tested on Pymolv0.97, Windows platform, Red Hat Linux 9.0 and Fedora Core 4. Will test v0.98 and MacOSX later on.
- The way it's setup now, when you import rotamers , it will automatically read-in the rotamer database; this may not be what you want.
- Post problems in the discussion page, on 'my talk' page or just email me : dwkulp@mail.med.upenn.edu
TASKS TODO:
- Rotamer Movie, using mset, etc create movie to watch cycle through rotamers
- Code could be organized a bit better; due to time constraints this is good for now..
TASKS DONE:
- Store crystal structure in rotamer menu, so you can go back to original orientation
USAGE
colorRotamers selection set_rotamer selection, chi1_angle [,chi2_angle] [,chi3_angle] [,chi4_angle] set_phipsi selection phi_angle, psi_angle createRotamerPBDs selection [,ncutoff] [,pcutoff] [,prefix]
EXAMPLES
colorRotamers chain A set_rotamer resi 40, -60,-40 (only set chi1,chi2 angles) set_phipsi resi 10-40, -60,-60 (create an alpha-helical-like section) createRotamerPDBs resi 10-12, ncutoff=3 (create 9 PDBs; each with one of the 3 most probable rotamers for resi 10,11,12) createRotamerPDBs resi 14, pcutoff=0.4 (create a pdb file for each rotamer of residue 14 with probablity > 0.4)
REFERENCES
Dunbrack and Cohen. Protein Science 1997
Dunbrack Lab Page (Contains backbone-dependent library)
SCRIPTS (Rotamers.py ; MyMenu.py)
Rotamers.py
##################################################################
# File: Rotamers.py
# Author: Dan Kulp
# Creation Date: 6/8/05
# Contact: dwkulp@mail.med.upenn.edu
#
# Notes:
# Incorporation of Rotamer library
# readRotLib() - fills rotdat;
# indexed by "RES:PHI_BIN:PSI_BIN".
#
# Three main functions:
# 1. colorRotamers - colors according
# to rotamer probablitity
# 2. getBins(sel)
# phi,psi bin for rotamer
# 3. set_rotamer - set a side-chain
# to a specific rotamer
#
# To setup a rotamer menu in the
# right click, under "Residue"
# 1. cp mymenu.py modules/pymol/menu.py
# 2. cp rotamers.py modules/pymol/rotamers.py (update ROTLIB)
#
# Requirements:
# set ROTLIB to path for rotamer library
# Reference:
# Dunbrack and Cohen. Protein Science 1997
####################################################################
import colorsys,sys
import editing
import os
import cmd
import math
# Path for library
ROTLIB=os.environ['PYMOL_PATH']+"/modules/pymol/bbdep02.May.sortlib"
# Place for library in memory..
rotdat = {}
def readRotLib():
# Column indexes in rotamer library..
RES = 0
PHI = 1
PSI = 2
PROB = 8
CHI1 = 9
CHI2 = 10
CHI3 = 11
CHI4 = 12
if os.path.exists(ROTLIB):
print "File exists: "+ROTLIB
input = open(ROTLIB, 'r')
for line in input:
# Parse by whitespace (I believe format is white space and not fixed-width columns)
dat = line.split()
# Add to rotamer library in memory :
# key format RES:PHI_BIN:PSI_BIN
# value format PROB, CHI1, CHI2, CHI3, CHI4
key=dat[RES]+":"+dat[PHI]+":"+dat[PSI]
if key in rotdat:
rotdat[key].append([ dat[PROB], dat[CHI1], dat[CHI2], dat[CHI3], dat[CHI4] ])
else:
rotdat[key] = [ [ dat[PROB], dat[CHI1], dat[CHI2], dat[CHI3], dat[CHI4] ] ]
else:
print "Couldn't find Rotamer library"
# Atoms for each side-chain angle for each residue
CHIS = {}
CHIS["ARG"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","CD" ],
["CB","CG","CD","NE" ],
["CG","CD","NE","CZ" ]
]
CHIS["ASN"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","OD2" ]
]
CHIS["ASP"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","OD1" ]
]
CHIS["CYS"] = [ ["N","CA","CB","SG" ]
]
CHIS["GLN"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","CD" ],
["CB","CG","CD","OE1"]
]
CHIS["GLU"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","CD" ],
["CB","CG","CD","OE1"]
]
CHIS["HIS"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","ND1"]
]
CHIS["ILE"] = [ ["N","CA","CB","CG1" ],
["CA","CB","CG1","CD1" ]
]
CHIS["LEU"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","CD1" ]
]
CHIS["LYS"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","CD" ],
["CB","CG","CD","CE"],
["CG","CD","CE","NZ"]
]
CHIS["MET"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","SD" ],
["CB","CG","SD","CE"]
]
CHIS["PHE"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","CD1" ]
]
CHIS["PRO"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","CD" ]
]
CHIS["SER"] = [ ["N","CA","CB","OG" ]
]
CHIS["THR"] = [ ["N","CA","CB","OG1" ]
]
CHIS["TRP"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","CD1"]
]
CHIS["TYR"] = [ ["N","CA","CB","CG" ],
["CA","CB","CG","CD1" ]
]
CHIS["VAL"] = [ ["N","CA","CB","CG1" ]
]
# Color Rotamer by side-chain angle position
# 'bin' side-chain angles into closest
def colorRotamers(sel):
doRotamers(sel)
# Utility function, to set phi,psi angles for a given selection
# Note: Cartoon, Ribbon functionality will not display correctly after this
def set_phipsi(sel, phi,psi):
doRotamers(sel,angles=[phi,psi],type="set")
# Set a rotamer, based on a selection, a restype and chi angles
def set_rotamer(sel, chi1, chi2=0,chi3=0,chi4=0):
at = cmd.get_model("byres ("+sel+")").atom[0]
list = [chi1,chi2,chi3,chi4]
for i in range(len(CHIS[at.resn])):
print "Setting Chi"+str(i+1)+" to "+str(list[i])
editing.set_dihedral(sel + ' and name '+CHIS[at.resn][i][0],
sel + ' and name '+CHIS[at.resn][i][1],
sel + ' and name '+CHIS[at.resn][i][2],
sel + ' and name '+CHIS[at.resn][i][3], str(list[i]))
# Remove some objects that got created
cmd.delete("pk1")
cmd.delete("pk2")
cmd.delete("pkmol")
# Get Phi,Psi bins for given selection
# WARNING: assume selection is single residue (will only return first residue bins)
def getBins(sel):
return doRotamers(sel, type="bins")
# Color Ramp...
def rot_color(vals):
nbins = 10
vals.sort(key=lambda x:x[1])
# print "End sort: "+str(len(vals))+" : "+str(nbins)
# Coloring scheme...
j = 0
rgb = [0.0,0.0,0.0]
sel_str = ""
for i in range(len(vals)):
if int(len(vals)/nbins) == 0 or i % int(len(vals)/nbins) == 0:
hsv = (colorsys.TWO_THIRD - colorsys.TWO_THIRD * float(j) / (nbins-1), 1.0, 1.0)
#convert to rgb and append to color list
rgb = colorsys.hsv_to_rgb(hsv[0],hsv[1],hsv[2])
if j < nbins-1:
j += 1
cmd.set_color("RotProbColor"+str(i), rgb)
cmd.color("RotProbColor"+str(i), str(vals[i][0]))
# Main function
def doRotamers(sel,angles=[], type="color"):
# Read in Rotamer library if not already done
if len(rotdat) == 0:
readRotLib()
# Set up some variables..
residues = ['dummy'] # Keep track of residues already done
probs = [] # probability of each residue conformation
phi = 0 # phi,psi angles of current residue
psi = 0
# Get atoms from selection
atoms = cmd.get_model("byres ("+sel+")")
# Loop through atoms in selection
for at in atoms.atom:
try:
# Don't process Glycines or Alanines
if not (at.resn == 'GLY' or at.resn == 'ALA'):
if at.chain+":"+at.resn+":"+at.resi not in residues:
residues.append(at.chain+":"+at.resn+":"+at.resi)
# Check for a null chain id (some PDBs contain this)
unit_select = ""
if at.chain != "":
unit_select = "chain "+str(at.chain)+" and "
# Define selections for residue i-1, i and i+1
residue_def = unit_select+'resi '+str(at.resi)
residue_def_prev = unit_select+'resi '+str(int(at.resi)-1)
residue_def_next = unit_select+'resi '+str(int(at.resi)+1)
# Compute phi/psi angle
phi = cmd.get_dihedral(residue_def_prev+' and name C',residue_def+' and name N',residue_def+' and name CA',residue_def+' and name C')
psi = cmd.get_dihedral(residue_def+' and name N',residue_def+' and name CA',residue_def+' and name C',residue_def_next+' and name N')
if type == "set":
print "Changing "+at.resn+str(at.resi)+" from "+str(phi)+","+str(psi)+" to "+str(angles[0])+","+str(angles[1])
cmd.set_dihedral(residue_def_prev+' and name C',residue_def+' and name N',residue_def+' and name CA',residue_def+' and name C',angles[0])
cmd.set_dihedral(residue_def+' and name N',residue_def+' and name CA',residue_def+' and name C',residue_def_next+' and name N', angles[1])
continue
# Find correct 10x10 degree bin
phi_digit = abs(int(phi)) - abs(int(phi/10)*10)
psi_digit = abs(int(psi)) - abs(int(psi/10)*10)
# Remember sign of phi,psi angles
phi_sign = 1
if phi < 0: phi_sign = -1
psi_sign = 1
if psi < 0: psi_sign = -1
# Compute phi,psi bins
phi_bin = int(math.floor(abs(phi/10))*10*phi_sign)
if phi_digit >= 5: phi_bin = int(math.ceil(abs(phi/10))*10*phi_sign)
psi_bin = int(math.floor(abs(psi/10))*10*psi_sign)
if psi_digit >= 5: psi_bin = int(math.ceil(abs(psi/10))*10*psi_sign)
print "Given "+at.resn+":"+at.resi+" PHI,PSI ("+str(phi)+","+str(psi)+") : bin ("+str(phi_bin)+","+str(psi_bin)+")"
# Get current chi angle measurements
chi = []
for i in range(len(CHIS[at.resn])):
chi.append(cmd.get_dihedral(residue_def + ' and name '+CHIS[at.resn][i][0],
residue_def + ' and name '+CHIS[at.resn][i][1],
residue_def + ' and name '+CHIS[at.resn][i][2],
residue_def + ' and name '+CHIS[at.resn][i][3]))
print "CHIs: "+str(chi)
if type == 'bins':
return [at.resn, phi_bin,psi_bin]
# Compute probabilities for given chi angles
prob = 0
prob_box = 22
for item in range(len(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)])):
print "Rotamer from db: "+str(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item])
if chi[0]:
if chi[0] >= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][1]) - (prob_box/2) and \
chi[0] <= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][1]) + (prob_box/2):
if len(chi) == 1:
prob = rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][0]
break
if chi[1] >= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][2]) - (prob_box/2) and \
float(chi[1] <= rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][2]) + (prob_box/2):
if len(chi) == 2:
prob = rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][0]
break
if chi[2] >= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][3]) - (prob_box/2) and \
float(chi[2] <= rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][3]) + (prob_box/2):
if len(chi) == 3:
prob = rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][0]
break
if chi[3] >= float(rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][4]) - (prob_box/2) and \
float(chi[3] <= rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][4]) + (prob_box/2):
prob = rotdat[at.resn+":"+str(phi_bin)+":"+str(psi_bin)][item][0]
break
print "PROB OF ROTAMER: "+str(prob)
print "---------------------------"
probs.append([residue_def, prob])
except:
# probs.append([residue_def, -1])
print "Exception found"
continue
# Color according to rotamer probability
rot_color(probs)
# Create PDB files containing most probable rotamers
def createRotamerPDBs(sel,ncutoff=10,pcutoff=0,prefix="ROTAMER"):
# Get atoms from selection
atoms = cmd.get_model("byres ("+sel+")")
# Set up some variables..
residues = ['dummy'] # Keep track of residues already done
# Loop through atoms in selection
for at in atoms.atom:
if at.resn in ('GLY','ALA') or "%s:%s:%s" % (at.chain,at.resn,at.resi) in residues:
continue
# Add to residue list (keep track of which ones we've done)
residues.append("%s:%s:%s" % (at.chain,at.resn,at.resi))
# Check for a null chain id (some PDBs contain this)
unit_select = ""
if not at.chain == "":
unit_select = "chain "+str(at.chain)+" and "
# Define selections for residue
residue_def = unit_select+'resi '+str(at.resi)
# Get bin (phi,psi) definitions for this residue
bin = doRotamers(residue_def, type='bins')
# Store crystal angle
crystal_angles = [0.0,0.0,0.0,0.0]
for angle in range(3):
try:
crystal_angles[angle] = bin[3][angle]
except IndexError:
break
# Retreive list of rotamers for this phi,psi bin + residue type
match_rotamers = rotdat["%s:%s:%s" % (bin[0],str(bin[1]),str(bin[2]))]
count = 0
for item in range(len(match_rotamers)):
# Store probablity
prob = match_rotamers[item][0]
# Check cutoffs
if float(prob) <= float(pcutoff):
continue
if float(count) >= float(ncutoff):
break
# Increment count
count += 1
# Output to screen ...
print "Residue %s%s, rotamer %i, prob %s" % (str(at.resn),str(at.resi),int(item),str(prob))
# Set to new rotamer
set_rotamer(residue_def,match_rotamers[item][1],match_rotamers[item][2],match_rotamers[item][3],match_rotamers[item][4])
# Store in PDB file
cmd.save("%s_%s%s_%i_%s.pdb" % (prefix,str(at.resn),str(at.resi),int(item),str(prob)))
# Reset crystal angle
set_rotamer(residue_def,crystal_angles[0],crystal_angles[1],crystal_angles[2],crystal_angles[3])
# Uncommenting this is nice because it loads rotamer library upon startup
# however, it slows the PyMOL loading process a lot
# instead I've put this call into the menuing code..
# readRotLib()
cmd.extend('set_phipsi',set_phipsi)
cmd.extend('set_rotamer',set_rotamer)
cmd.extend('colorRotamers',colorRotamers)
cmd.extend('createRotamerPDBs',createRotamerPDBs)
MyMenu.py
Since menu.py is copyrighted I can't post my edited version, but you can create it very simply by adding these two peices of code
1. In the "pick_option(title,s,object=0)" function of menu.py add the following code after the first "result =" statement
# Edit dwkulp 6/11/05 , add a rotamer menu to residue menu
if title == 'Residue':
result.extend([[ 1, 'rotamers' , rotamer_menu(s)]])
2. At the end of the file add this:
###############################################
# Dan Kulp
# Added Rotamer list to residue menu..
# rotamer.py must be importable (I placed it in
# the same directory as menu.py)
###############################################
import rotamers
def rotamer_menu(s):
# Check for rotamer library being loaded
if not rotamers.rotdat:
rotamers.readRotLib()
# return [ [2, "Must run rotamers.py first",'']]
# Check for valid rotamer residue..
res = cmd.get_model("byres ("+s+")").atom[0].resn
if not res in rotamers.CHIS.keys():
return [ [2, "Residue: "+res+" not known sidechain or does not have rotamers", '']]
# Get PHI,PSI bins for rotamer (also prints out current phi,psi, chi1,chi2,chi3,chi4)
bins = rotamers.doRotamers(s,type='bins')
# Add a title to the menu
result = [ [2, bins[0]+' Rotamers in bin ('+str(bins[1])+','+str(bins[2])+')','' ], [1, ':::PROB,CHI1,CHI2,CHI3,CHI4:::','']]
# Grab the entries for this residue and phi,psi bins
match_rotamers = rotamers.rotdat[bins[0]+":"+str(bins[1])+":"+str(bins[2])]
# Set max number of rotamers to display (probably should be somewhere 'higher up' in the code)
max_rotamers = min(10, len(match_rotamers))
# Create menu entry for each possible rotamer
for item in range(max_rotamers):
result.append( [ 1, str(match_rotamers[item]), 'rotamers.set_rotamer("'+s+'","'\
+str(match_rotamers[item][1])+'","'\
+str(match_rotamers[item][2])+'","'\
+str(match_rotamers[item][3])+'","'\
+str(match_rotamers[item][4])+'")'])
return result