This is a read-only mirror of pymolwiki.org
Difference between revisions of "Colorbydisplacement"
m (18 revisions) |
|||
| (8 intermediate revisions by one other user not shown) | |||
| Line 1: | Line 1: | ||
| − | == | + | {{Infobox script-repo |
| − | + | |type = script | |
| − | + | |filename = colorbydisplacement.py | |
| − | + | |author = [[User:Tlinnet|Troels E. Linnet]] | |
| + | |license = BSD | ||
| + | }} | ||
== Introduction == | == Introduction == | ||
This script allows you to color two structures by distance displacement between an Open and Closed form of a protein, as calculated by PyMol's internal distance command. The pairwise distance is calculated between all-atoms. The distance displacement values are stored as B-factors of these residues, which are colored by a ''rainbow'' color spectrum, with blue specifying minimum and red indicating maximum. | This script allows you to color two structures by distance displacement between an Open and Closed form of a protein, as calculated by PyMol's internal distance command. The pairwise distance is calculated between all-atoms. The distance displacement values are stored as B-factors of these residues, which are colored by a ''rainbow'' color spectrum, with blue specifying minimum and red indicating maximum. | ||
| − | |||
Do keep in mind, all original B-factors values are overwritten! | Do keep in mind, all original B-factors values are overwritten! | ||
| Line 37: | Line 38: | ||
Residues not in both pdb files is colored black | Residues not in both pdb files is colored black | ||
| − | == Example | + | == Example 1 == |
| − | + | {{Template:PymolScriptRepoDownload|examples/colorbydisplacement_1.pml}} | |
| − | + | <include src="https://raw.github.com/Pymol-Scripts/Pymol-script-repo/master/examples/colorbydisplacement_1.pml" highlight="python" /> | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | ||
| − | |||
| − | = | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
[[Category:Script_Library]] | [[Category:Script_Library]] | ||
[[Category:Structural_Biology_Scripts]] | [[Category:Structural_Biology_Scripts]] | ||
| + | [[Category:Pymol-script-repo]] | ||
Latest revision as of 02:15, 28 March 2014
| Type | Python Script |
|---|---|
| Download | colorbydisplacement.py |
| Author(s) | Troels E. Linnet |
| License | BSD |
| This code has been put under version control in the project Pymol-script-repo | |
Introduction
This script allows you to color two structures by distance displacement between an Open and Closed form of a protein, as calculated by PyMol's internal distance command. The pairwise distance is calculated between all-atoms. The distance displacement values are stored as B-factors of these residues, which are colored by a rainbow color spectrum, with blue specifying minimum and red indicating maximum.
Do keep in mind, all original B-factors values are overwritten!
There exist one version.
ColorByDisplacementAll is between All atoms in residues and is quite slow => 3-5 mins for a run. Ideal for sticks representation.
You have to specify which residues should be used in the alignment procedure, or it will take all residues as standard
V.2 is implemented the 2011.01.06 - Due to a bug in coloring.
Bug in code
A bug in the boolean operator of the spectrum command has been found. This versions work for version 1.3 Educational product.
For other versions of pymol, try to change (comment/uncomment) the cmd.spectrum line.
The other spectrum line works for Open-Source PyMOL 1.2r3pre, Incentive product
Examples
ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t
ColorByDisplacementAll O5NT, C5NT, super1=resi 26-355, super2=resi 26-355, doColor=t, doAlign=t, AlignedWhite='no'
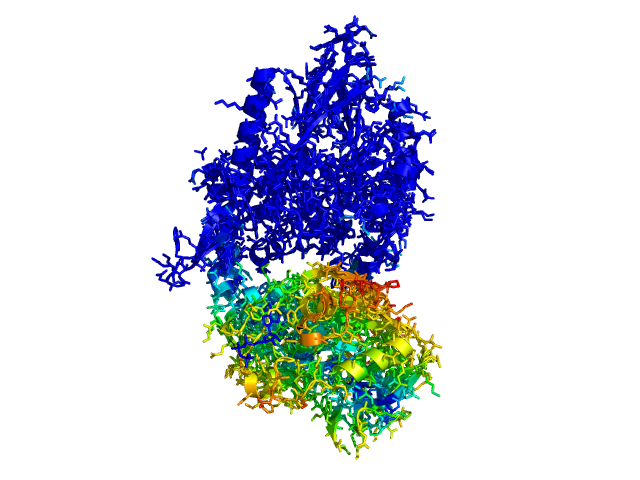
ColorByDisplacementAll used on 1HP1 and 1HPU aligned and colored by distance displacement.
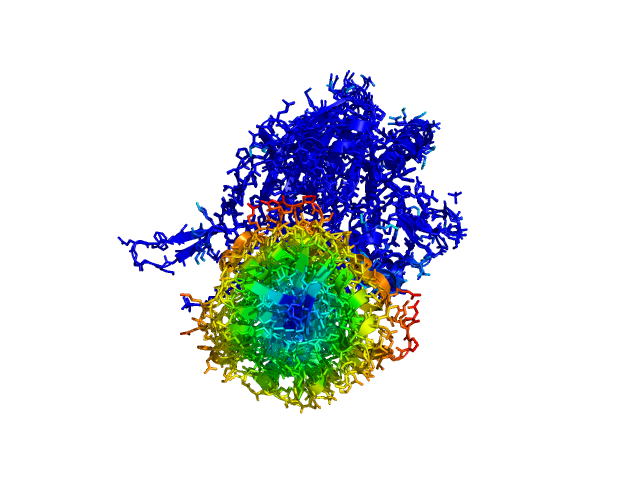
ColorByDisplacementAll used on 1HP1 and 1HPU aligned and colored by distance displacement.


Dark blue is low displacement, higher displacements are in orange/yellow/red.
Residues used for alignment is colored white. Can be turned off in top of algorithm.
Residues not in both pdb files is colored black
Example 1
| Download: examples/colorbydisplacement_1.pml | |
| This code has been put under version control in the project Pymol-script-repo | |
<include src="https://raw.github.com/Pymol-Scripts/Pymol-script-repo/master/examples/colorbydisplacement_1.pml" highlight="python" />